
-
药业专题
 展会信息
展会信息
联系我们
地址:广州市越秀区大沙头路东四巷16号四楼
电话:020-87301042
传真:020-87303289
Email:gdzyxh@zkgroup.info
-

新闻动态
- 您现在的位置:首页 > 新闻动态
政策法规改革对中国医药及创新药的影响
与药品研发(R&D)领域的科学创新相似,在过去的二十年中,中国通过监管创新对药品法规进行了重大变革,以加快药品审评审批。而中国创新药物的蓬勃发展,尤其受益于支持创新的政策。统计研究发现,从2005年到2021年,随着具有新作用机制的药物出现,包括免疫检查点抑制剂和细胞治疗产品,进口和国产新型抗癌药物的数量都显著增加。
与2006-2010年相比,2016-2020年进口药品的药品滞后期也大幅缩短了70%以上。最近,来自于NPMA和清华大学等机构的学者发文详细分析了中国抗癌药法规演变与监管创新,以及其对中国抗癌药物和创新药物开发的影响。
NMPA由来
自成立以来,中国的药品监管机构在过去的二十年中不断发展。1998年,国家药品监督管理局(SDA)成立,以解决跨省药品审批标准不一致问题,这标志着法律制度的开始(图1)。1999 年,SDA 首次发布并根据需要进行修订的《中国临床试验质量管理规范指南》确立了临床研究的质量和完整性标准。2003年,食品行政管理职能并入国家食品药品监督管理局,由该局转变为国家食品药品监督管理局。随后的国家食品药品监督管理总局(CFDA)成立于2013年,升格为国务院部级机构。
2018 年,作为中国政府行政改革的一部分,CFDA 成为新成立的国家市场监督管理总局下属的“国家药品监督管理局”(NMPA)。药品审评中心(CDE)隶属于国家药监局及其前身,负责药品(包括化学、生物制品和中药)的临床试验申请、上市许可申请以及后续修订和续展的科学评估。为了使其结构更符合现代化医药,CDE实施了多轮机构重组,以简化审查程序。到 2016年底,CDE的员工数量翻了两番,并且进一步扩大,这也为更高效的审查流程做出了巨大贡献。通过采取一系列监管创新来加速药物开发和药物审评,监管机构正在成为一个以科学为基础、以临床价值为基础的监管机构,旨在确保药物的有效性、安全性和质量。鉴于与各种恶性肿瘤相关的未得到满足的医疗需求不断上升,抗癌药物一直是药物研发 (R&D) 的重点,它代表了大多数加快项目指定的药物,并在中国新批准的药物中占据主导地位。
在这里,来自于NMPA和清华大学的研究者统计分析了2005年至2021年中国抗癌药物批准的趋势和特征,以说明监管创新对药物研发的影响。
 图1:Iteration of China’s key pharmaceutical laws and policies since 1998
图1:Iteration of China’s key pharmaceutical laws and policies since 1998
关键法律和政策的迭代
中国的药品监管框架由多个层次组成。规范药品(包括生物制品)的主要法规是《中华人民共和国药品管理法》(DAL),并辅以一套通用实施细则,简称《药品管理法实施条例》。DAL之下,药物注册条例 (DRR) 是管理临床试验和药物注册的核心文件。另外, CDE 同样参与起草与药物开发和注册相关的指导文件。在过去的几十年中,针对历史事件,立法和政策进行了重大的迭代和调整。监管法规最初基于第一版《药品管理法》(DAL,1984 年颁布,2001 年修订)和 DRR(2002年颁布,2005年生效)。2007年,在发生多起药物事故后,如鞘内注射被长春新碱污染的甲氨蝶呤引起的严重神经系统不良反应,当局对 DRR 进行了实质性修改,以加强对药品质量的监管。在接下来的十年中,直到2015年都没有对DAL和DRR进行进一步的全面修订。
2015年,随着公众健康需求与审批步伐缓慢的矛盾不断加剧,监管改革的号角开始吹响。在标志性文件《关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号)和《关于深化审评审批制度改革鼓励创新的意见》,及《药品医疗器械管理办法》(国发〔2017〕42 号)的指导下,一系列前所未有的举措出台,用于提高药品审评效率、激发临床价值创新、提升药品质量等。为了与监管创新的实施相结合,CFDA 于2017年作为监管成员加入国际协调委员会 (ICH),并承诺在中国实施国际标准。在鼓励创新药的同时,监管机构也鼓励仿制药,同时通过提高仿制药质量一致性评价要求规定的技术标准来强调药品质量。
为适应药品研发的快速发展,国家药监局逐步采用以临床价值为基础的药品监管体系。升级后的系统现在正在引领行业重视创新并关注患者的需求。DAL和 DRR的最新修订版(分别于 2019 年 12月1日和 2020年7月1日生效)纳入并优化了经过时间考验和有效的监管流程,包括快速指定程序、与监管机构的沟通机制以及 60个工作日的默许(IND)通过程。越来越多的科学和技术指南已经发布,以辅助新修订的DAL和DRR,而且在2021年,CDE更是创纪录的发布了87条指南。值得注意的是,最近颁布的一项指南强调了抗癌药物研发中“临床价值”的驱动力,这为以患者为中心的药物开发奠定了基础。此外,中国国家卫健委发布了关于药物安全性、有效性、经济价值、创新性、适宜性和可及性的全面、科学的药物评价框架试点指南。
中国获批的抗癌药物
2005年1月至 2021年5月,中国当局批准了660种抗癌药物,其中包括 94 种新药(表 1)。2005-2007年期间,绝大多数(241/258)为仿制药,13种新药上市。2008年,受多起药品事故和腐败案件的触发,主管部门决定加强药品管理和监管,对仿制药实行集体审评。2008 年的集体审查和 2016年的GQCE要求是显著提高仿制药标准和排除多余仿制药的两个最重要的里程碑。结果,多个临床申请被申办者自愿撤回。此外,注册积压影响了新药和仿制药的批准。在2017年之前,每年批准的抗癌药物数量显著下降。
特别是在2014-2017年期间,仅批准了13种新药和31种仿制药(表1)。随着监管改革和研发能力的提高,中国自2018 年以来出现了药物开发的爆炸式增长(图2A和 B,表1)。值得注意的是,2018年至 2021 年5月,获批的新药数量达到54个(33.5%)(表 1)。同期获批仿制药99个,生物类似药6个,改良新药2个。同样,在研的候选药物的数量在近几年也显著增加。
作为药物研发中的监管限速步骤,IND和NDA审查时间大大缩短。根据 2013年CDE 年度报告24,化学品和治疗药物IND申请的平均等待期分别为6个月和13个月。自从实行60个工作日的默许审批制度以来,据报道,2021年 99.86% 的IND申请在时限内获得批准。在新药申请 (NDA) 审评时长方面,抗癌新药申请时长的中位数从2008-2010年的18.7(17.5-25.6)个月下降到 2018年1月-- 2021年5月的12.5(10.0-17.2,P=0.0028)(下图A)。2008-2011年提交IND申请的新药获得上市许可的总中位时间为 8.1(7.3-9.1)年,而2012–2015年提交IND申请的该时间减少到 5.3(4.8-5.9,P<0.0001)年(下图 C)。
不断增长的科技创新促进了具有多种作用机制(MOA)的药物的开发(表1)。2010年之前,中国获批的抗癌药大多为细胞毒化疗药物,且以仿制药居多。在接下来的十年中,细胞毒药物的数量有所下降,而其他 MOA 的药物数量却在增加。具体而言,新靶向药物的数量一直在增长,从 2005-2007 年的5个产品(38.5%)增加到 2014-2017 年的11个产品(84.6%),并在2018年至 2021 年5月期间达到 39个产品(72.2%)。随后 ,靶向仿制药和生物仿制药最近显示出快速增长,2018 年至 2021 年5月批准了40种仿制药和6种生物仿制药。免疫疗法在 2018 年开始蓬勃发展,截至 2021 年5月有8种产品获批。值得注意的是,两种嵌合抗原受体 (CAR) -T细胞疗法于分别于2021年6月和2021年9月在中国获批。
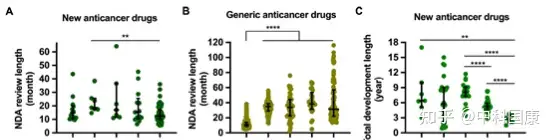 图2:Trend of NDA review length and total development length of new anticancer drugs approved in China, 2005 to 2021
图2:Trend of NDA review length and total development length of new anticancer drugs approved in China, 2005 to 2021
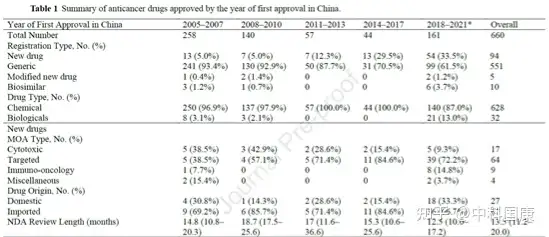 表1:Summary of anticancer drugs approved by the year of first approval in China.
表1:Summary of anticancer drugs approved by the year of first approval in China.
推动国产药物研发
自2007年以来,中国制定了多个加速项目,用以不断促进国内创新。第一个行动是在行业和监管机构之间建立沟通渠道。特别审查 (SR) 计划最初是在2007年修订的DRR中引入的,用于治疗严重疾病并显示出实质性临床益处的新药(图1,表2)。具有SR称号的药物将在 CDE 的开发和注册方面得到更严格的指导。研究发现,39种(41.5%)获批的新抗癌药物被授予SR称号(图 3)。
SR指定侧重于沟通,但对NDA审查长度和总开发长度几乎没有影响(图4)。自 2016年发起人与监管机构之间建立正式沟通渠道后,该计划的好处变得不那么明显。因此,SR指定在2020年终止。2015年12月,作为全面监管改革的先驱措施之一,优先审评(PR)项目,可以有效的缩短药品审评时间。PR计划的目标最初是为了减少注册积压,随后在 2017年12月转向提高临床价值,可以解决未满足的医疗需求并显示出大量临床益处的药物有资格获得这一称号。2017年后批准的18个(100%)国产抗癌新药获得PR(图3),中位审评时间为14.6个月(图4)。
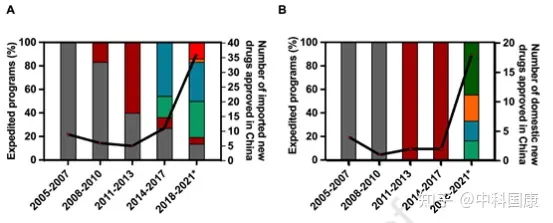 图3:Uptake of expedited programs (EP)
图3:Uptake of expedited programs (EP)
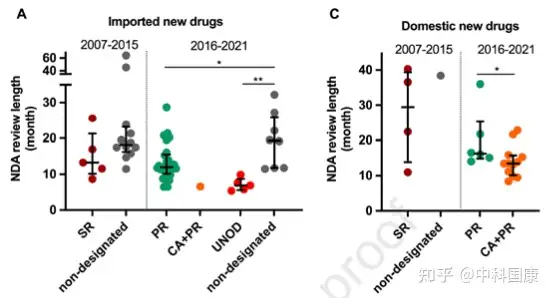
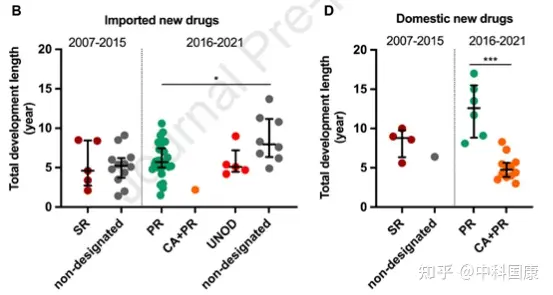 图4:NDA review length and total development length of approved new anticancer drugs in China by expedited programs, 2007 to May 2021
图4:NDA review length and total development length of approved new anticancer drugs in China by expedited programs, 2007 to May 2021
监管机构引入了两个额外的计划来增加监管灵活性,包括有条件批准 (CA) 和突破性治疗 (BT)(表 2)。2017 年12月,基于可合理预测临床益处或不太全面的临床数据的替代终点建立了用于批准药物的CA途径。CA适用于在危及生命或罕见疾病的情况下,临床价值高于现有疗法的药物。例如,与第一代抑制剂依鲁替尼相比,可能具有疗效和安全性优势的两种国产新一代布鲁顿酪氨酸激酶 (BTK) 抑制剂 zanubrutinib 和 orelabrutinib 获得 NMPA 有条件批准用于复发/难治性慢性淋巴细胞白血病患者或小淋巴细胞性淋巴瘤。
与美国的加速批准计划类似,在中国接受CA的药物需要在上市后继续确认其实际临床益处。统计结构表明,CA 指定与减少的总开发时间长度显著相关(图 4D)。12种国内PR指定药物在CA途径下获得批准,IND 提交后的中位数为 4.8 (3.9-5.7) 年,而国内PR指定药物未接受CA的时间为 12.6 (8.9-15.5) 年(差异,7.8 年; P = 0.0002)。BT是在2020 年修订的DRR中首次引入的称号,该称号给予初步临床数据显示比现有疗法有很大的临床益处的药物。BT 指定药物的开发可以通过与当局的密切沟通来加快开发进程,这些药物可能有资格获得CA和PR途径。截至 2022年5月,46种抗癌新药获得中国BT认定。其中5款已获 NMPA 批准用于 相关适应症。
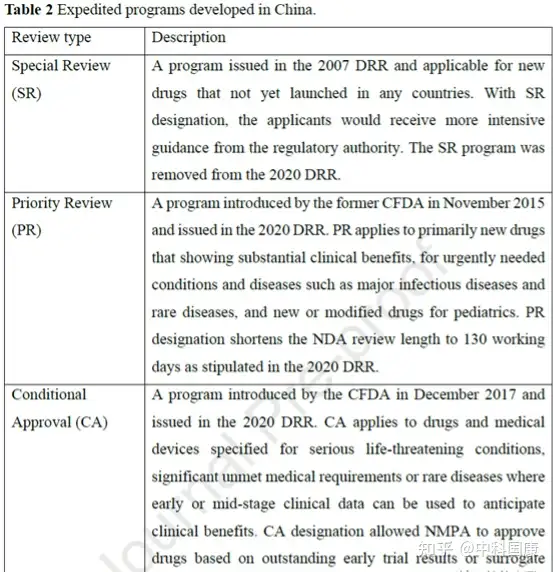 表2:Expedited programs development in china
表2:Expedited programs development in china

减少药物滞后
促进进口新药的可及性是解决未满足的医疗需求的另一种方式。在 2000年之前,当确定持久的医疗需求时,无需在中国进行额外的临床试验即可批准药物。随着研发和监管经验的积累,当局开始要求在上市批准之前提供中国数据。2017年之前,对于新药而言,在海外开展2期试验是中国机构参与多中心临床试验的前提,在中国上市前必须持有其他国家颁发的上市许可证。跨国制药公司因此通过桥接研究将全球批准的药物引入中国,该政策在2008年至2013年期间主导了中国的注册策略。
此外,低效的药物审评过程是进口药物进入的另一个障碍。调查结果表明,2008-2013年进口新药的NDA审评时间最长,为17.7(15.9-22.6)个月。正如 2013 年CDE年度报告所报告的那样,NDA审查的积压情况越来越多(长达14 个月的等待期),而进行桥接研究的化学药物IND审查的等待期甚至更长(长达 19 个月)。因此,中国的药物研发和批准显著落后于西方,2001-2005年在美国开始临床试验的抗癌药物在中国滞后5.8(3.4-7.8)年进入临床阶段;) 2006 -2010年,在美国批准的抗癌药物进入中国滞后6.6(2.8-9.5)年。这种药物滞后(定义为一种新药在美国获批和随后在中国获批之间的时间)限制了药物的可及性并阻碍了药物的创新。
 图5:Clinical development and first approval of imported new anticancer drugs inthe US and China
图5:Clinical development and first approval of imported new anticancer drugs inthe US and China自2015年以来,中国监管部门采取了多项开创性措施来改善中国的监管和研发环境(图 1)。并且自2017年10月起,监管结构允许在中国参与者中对在中国境外开发的新药物进行同步的首次人体研究(图 1)。此外,2017年加入ICH为中国注册技术要求与全球标准的统一铺平了道路。这些措施有利于中国早期参与多中心临床试验,促进患者可及性。随后,采用 MRCT 途径的药物比例开始扩大,在 2015年至2021年5月期间达到52%。与桥接临床实验相比,采用MRCT的药物可以缩短中美之间药物滞后的问题(3.8 VS 4.7 年,P = 0.225)。此外,中国一直在简化审批程序, PR指定新药的审评时间大大缩短。对于 31/44 指定为 PR 的进口药物,NDA审查时间中位数为12.0(10.2-15.5)个月,而非指定药物为19.4(11.8-25.9,P = 0.040)个月(图 4A)。这也伴随着相对较短的总开发时长(PR,5.7 [5.0-7.5] 年与非指定PR的8.0 [6.4-11.2] 年;图 4B)。
2018年国家药监局发布的指南正式采纳了境外试验临床数据。2018年至2020年发布的加快批准外海外急需药品(UNOD)清单就是一个典型的例证。UNOD 指定的药物通常具有显著的临床益处,并且如果没有种族差异,则可以在有限甚至没有中国临床数据的情况下申请上市许可,这可以有效缩短 NDA/生物许可申请 (BLA) 审查时间三到六个月。UNOD 名单上的五种抗癌药物在IND提交后上市的中位时间为 5.1(4.5-7.2)年(图 4B),中位审查时间为 6.9(5.6-8.8)个月(图 4A)。其中,2018年获美国食品药品监督管理局(FDA)批准的FLT3抑制剂gilteritinib,与挽救性化疗相比,显著提高了FLT3突变的复发或难治性(r/r)急性髓细胞白血病患者的生存率。鉴于中国这些患者的治疗选择有限,gilteritinib 于2020年11月被列入UNOD清单,并于2021年2月获得NMPA批准。
由于制药行业、临床和监管机构的共同努力,近年来药物滞后时间明显缩短。伴随着药物批准差距的缩短(1.9 [1.0-2.9] 年,2016-2020年 vs. 6.6 [2.8-9.5] 年,2006-2010年;P = 0.0006,图5),2011-2015年在美国进入临床阶段的抗癌药物进入临床阶段的滞后时间急剧下降至 2.0 (1.4-3.5)年(相比之下,2001-2005 年为5.8 [3.4-7.8]年,P = 0.0009 )。拟合的 LOWESS 曲线显示了药物滞后减少的趋势,到2021年接近1.4年(图6)。因此,在过去的二十年中,美国获得批准的新型抗癌药物在中国获得许可的比例一直在上升(图7A)。截至2021年5月,2000年至2020年间在美国获得许可的抗癌药物中,有40.8%(69/169)已在中国获得上市许可,另有10.7%正在等待提交的 NDA/BLA审查(图 7B)。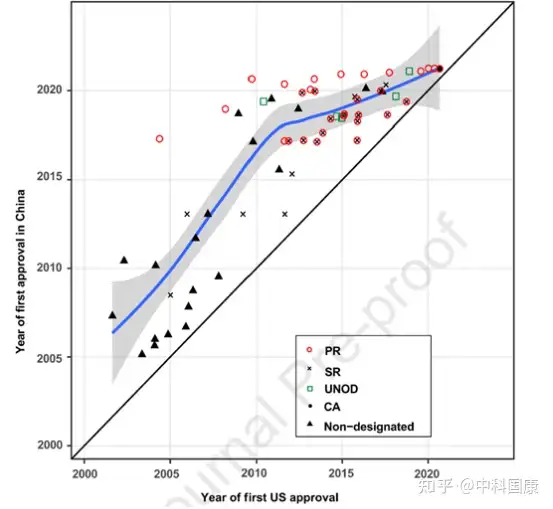 图6:First approval dates of imported new anticancer drugs in the US and China
图6:First approval dates of imported new anticancer drugs in the US and China
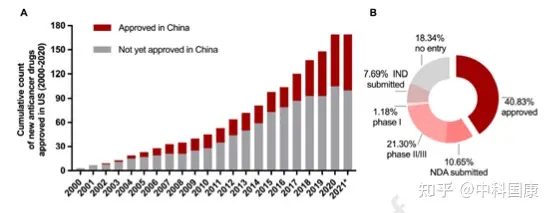 图7:Current status in China of new anticancer drugs approved in the US
图7:Current status in China of new anticancer drugs approved in the US
讨论
经过二十多年的改革,中国的药品监管机构已经升级为一个更加高效、科学和以临床价值为基础的体系,以跟上快速发展的科技创新步伐。我们见证了越来越多的进口和国产抗癌新药在中国获批,这在很大程度上得益于近期的监管变革、研发能力和技术创新。改善药物可及性以解决未满足的医疗需求一直是中国监管机构和药物开发商的首要任务。减少进口新药的滞后是关键一步。如图所示,与美国相比,药品滞后明显减少,尽管进口药品仍存在两年滞后。
改善药物可及性以解决未满足的医疗需求一直是中国监管机构和药物开发商的首要任务。减少进口新药的滞后是关键一步。如上文所述,尽管中国的进口药品与美国相比仍存在两年的进口滞后,但与美国相比,药品滞后显著减少。2020年最新修订的DRR规定的并行药物审查和基于风险的现场检查预计将进一步缩短审查时间。Pralsetinib是平行审查的一个例子,在PR和CA指定下审查长度为6.6个月。此外,向国内企业授权药品已成为海外生物科技企业抢先进入中国市场的有效途径。例如,Tesaro于2016年授予再鼎的niraparib 在 FDA 首次批准后的2.8年获得 NMPA批准。Axicabtagene ciloleucel是一种CD19导向的CAR-T细胞疗法,是2021年6月在中国获批的首个CAR-T细胞疗法,其技术是从Kite Pharma转移过来的YESCARTA®(axicabtagene ciloleucel)。
近年来,本土创新蓬勃发展,但多数是一些 “me too”类药物。事实上,适度的竞争对于卫生系统控制成本和提高药品负担能力是必要的,并且对于管理出现的药品短缺也至关重要。另外,患者也可以从一系列价格合理的抗癌药物中受益,包括me too、仿制药和生物类似药。然而,类似药物的重复开发会导致资源分配不当和对缺乏足够药物的治疗领域的投资不足。例如,中国已批准十余种抗 PD-1 或 PD-L1 单克隆抗体,但仍有大量类似的候选药物处于临床阶段。在仿制药中已经观察到相似之处,如中国市场上 60 多种阿奇霉素片的仿制药版本,而即使在参考药物的专利到期后,也没有仿制药硫酸阿扎那韦可用。药物开发的不平衡一直是一个全球性问题。为了应对这些障碍,FDA提供了一揽子监管激励措施,例如指定孤儿药,并建立了药物短缺数据库以监测药物可及性。这些集体努力使美国在急需领域获得批准的药物越来越多,包括数百个孤儿药和涉及先前短缺药物的仿制药批准。同样,中国当局发布了监管指南,强调未满足的医疗需求,并公布了几份专利过期或接近专利到期且没有仿制药的药品清单。未来,在采购、医院使用和定价方面,需要一个全面的价值和质量评估框架,这将鼓励开发具有实际临床价值和高质量仿制药的创新药。
中国制药行业目前正处于从“me-too”到“me-better”药物的过渡期。基于药物结构优化或基于先进技术平台的新药将表现出更好的疗效或安全性,或为患者提供替代益处。例如,世界上第一个皮下给药的 PD-L1 抗体 envafolimab 可能比静脉给药的 PD-(L)1 抗体显示出依从性和安全性优势。
事实上,一流的创新不可避免地会给发起人和监管机构带来更大的不可预见的风险。适当的风险管理和可靠的收益风险评估对于主动应对这些挑战至关重要。令人鼓舞的是,中国的监管体系不断适应外部变化,做出科学的监管决策。例如,康方生物自主研发的“全球首创”双特异性PD-1/CTLA-4抗体--卡多尼单抗,基于其良好的初步数据获得中国CDE的BT认定,与来自其他PD-1+CTLA-4 抗体联合疗法数据相比,在晚期宫颈癌的具有更高的客观缓解率。基于这些优异的结果,该药物已于 2022年6月通过加速途径获得批准。这反映了中国公司在开发 FIC 药物方面取得的进展,以及审评人员在对具有新作用机制的药物进行利益风险评估方面取得的进展。
全球化正在成为中国药物开发的常态,除了将中国纳入其全球注册战略的海外制药公司,本土公司也开始走向国际舞台。2020年,在中国开发的抗癌产品中约有五分之一也在境外开发。成功的全球发展需要严谨而前瞻性的战略规划,使用仅限中国的数据来支持其他国家的药物营销不可避免地具有挑战性。例如,FDA 已经对信迪利单抗和索凡替尼发出了完整的回复信。在做出监管决定时,FDA 曾表示,未满足的医疗需求程度和研究药物的临床获益程度将决定监管的灵活性程度,这与中国的监管机构的观点一致。值得注意的是,zanubrutinib 为第一个实现跨国同步开发的国内创新药物,获得了FDA和NMPA 的双重批准,并且其使用的关键数据主要来自中国患者。第一个本土开发的ADC药物disitamab vedotin 获得FDA的BT指定,用于HER2阳性局部晚期或转移性尿路上皮癌的二线治疗。作为该适应症的一种新颖且有前景的治疗方法,disitamab vedotin 满足了美国未满足的医疗需求。中国的NMPA一直在推动基于临床价值的药物开发,要求使用可用的最佳护理标准作为对照,并鼓励及时就关键试验设计和重大决策进行沟通。NMPA还致力于改进良好生产规范,并于去年被接受为药品检验合作计划的预加入成员。未来,同步药物开发和联合监管审查将进一步加速药物研发的全球化。
在整个药物开发过程中,全球多个监管和卫生社区一直在积极与患者互动。在美国,FDA 的以患者为中心的药物开发 (PFDD) 计划被纳入2012年重新授权的处方药用户费用法案 (PDUFA V) 和 2016颁布的21世纪治愈法案。FDA在实施有关患者参与的基本目标方面取得了重大进展,包括将患者体验数据纳入审查过程并提供PFDD指南。建立了“Project Patient Voice”等新型网络平台,以自愿报告患者的症状和副作用,补充临床试验数据的安全信息。在中国,以患者为中心将成为常态,近期肿瘤学研发指南中关于设计适合患者需求的临床试验和关于患者参与药物研发的指南都强调了这一点。此外,中国还一直在采用分散临床试验(DCT)的方法来减轻试验参与的负担,使试验能够连续进行,监管文件不断形成,并对DCT要素提供更明确的指导。